- 11 March 2020
- Posted by: nemcatgroup
- Category: Publications
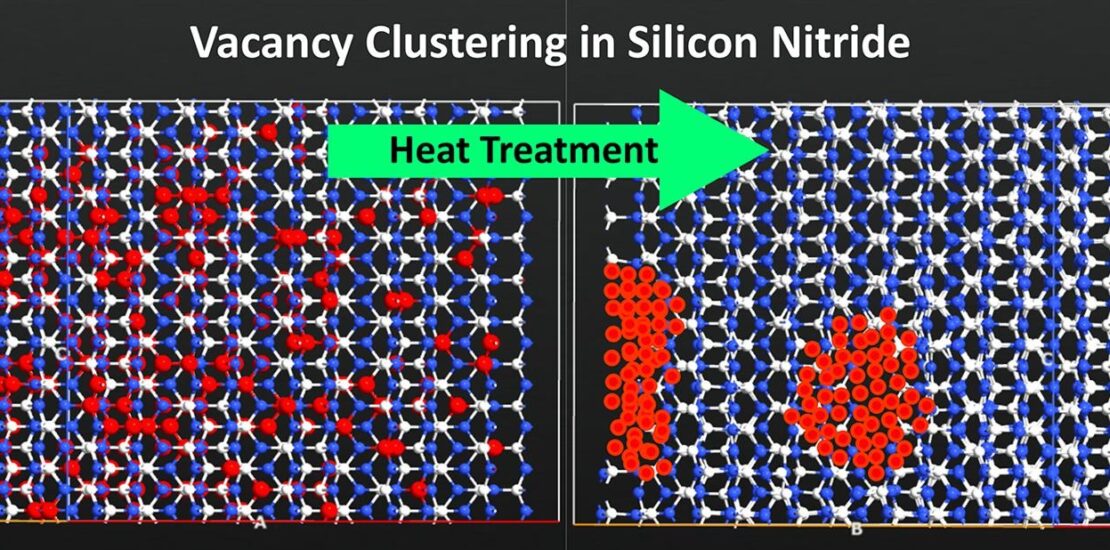
Molecular dynamics simulation is used to study vacancy cluster formation in β- and α-Si3N4 with varying vacancy contents (0–25.6 at%). Vacancies are randomly created in supercells, which were subsequently heat-treated for 114 ns. The results show that both β and α can tolerate vacancies up to 12.8 at% and form clusters, confirming previous experimental data indicating 8 at% vacancy in α-Si3N4. However, 25.6 at% vacancy in β results in complete amorphization, while the same amount in α results in a transformation of a semi-amorphous α phase to a defective β phase, leading to the removal of the clusters in newly formed β. This clearly explains why cluster vacancies are not experimentally observed in β, considering that β-Si3N4 ceramics are produced from α. Furthermore, the lattice parameters of both modifications increase with increasing vacancy content, revealing the cause of different lattice constants that were previously reported for α-Si3N4.